Getting Started with phylobuild
Kamil Slowikowski
2026-01-27
Source:vignettes/phylobuild.Rmd
phylobuild.RmdIntroduction
phylobuild provides fast neighbor-joining algorithms for phylogenetic tree construction from distance matrices. It wraps the highly optimized decenttree C++ library to bring substantial performance improvements over traditional implementations.
For large datasets (>1000 taxa), phylobuild can be 10-100x
faster than ape::nj().
Quick Start
The main function is fast_nj(), which works just like
ape::nj() but faster:
library(phylobuild)
library(ape)
# Load example primate data (dist object, Table 4 from Hayasaka et al. 1988)
data(primates)
as.matrix(primates)[1:5,1:5]
#> Human Chimpanzee Gorilla Orangutan Gibbon
#> Human 0.000 0.094 0.110 0.179 0.205
#> Chimpanzee 0.094 0.000 0.113 0.192 0.214
#> Gorilla 0.110 0.113 0.000 0.189 0.215
#> Orangutan 0.179 0.192 0.189 0.000 0.211
#> Gibbon 0.205 0.214 0.215 0.211 0.000Build a tree with the default RapidNJ algorithm:
tree <- fast_nj(primates)
tree
#>
#> Phylogenetic tree with 12 tips and 10 internal nodes.
#>
#> Tip labels:
#> Human, Chimpanzee, Orangutan, Gibbon, Japanese_macaque, Rhesus_macaque, ...
#>
#> Unrooted; includes branch length(s).The result is a standard phylo object compatible with
all ape functions:
plot(tree, type = "unrooted", lab4ut = "axial")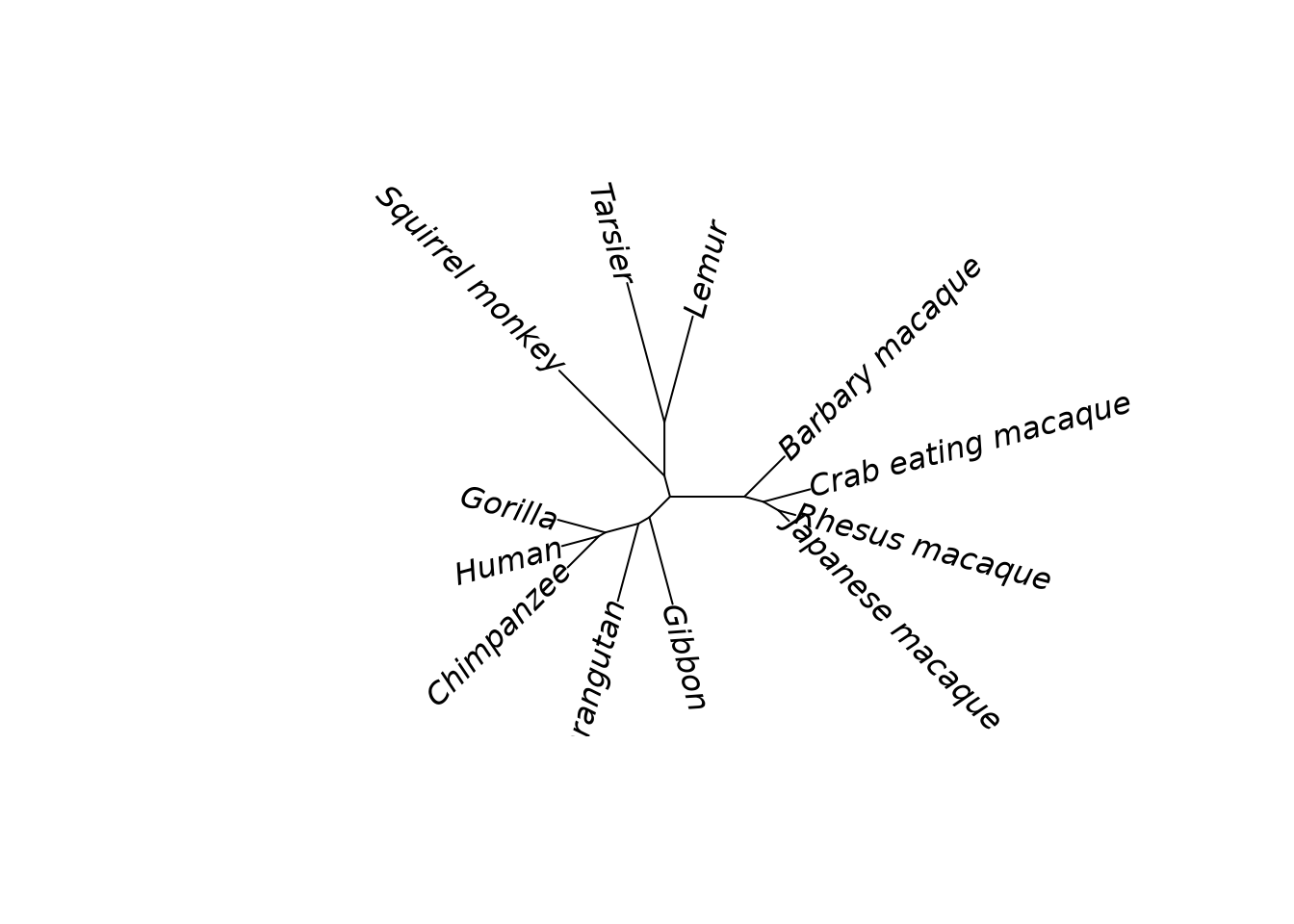
Primate phylogeny reconstructed with RapidNJ
Available Algorithms
phylobuild provides four neighbor-joining variants:
list_algorithms()
#> [1] "rapidnj" "nj" "bionj" "rapid_bionj"| Algorithm | Description | Best For |
|---|---|---|
rapidnj |
RapidNJ with branch-and-bound | Large datasets (default) |
nj |
Classic NJ (Saitou & Nei 1987) | Small datasets, exact NJ |
bionj |
BIONJ with variance estimates | When variance matters |
rapid_bionj |
RapidNJ + BIONJ | Large datasets + variance |
Comparing Algorithms
All algorithms produce similar trees, but with subtle differences:
tree_rapidnj <- fast_nj(primates, method = "rapidnj")
tree_nj <- fast_nj(primates, method = "nj")
tree_bionj <- fast_nj(primates, method = "bionj")Compare tree topologies using the Robinson-Foulds distance (0 = identical topology):
Performance Benchmarks
The key advantage of phylobuild is speed. Let’s compare
fast_nj() with ape::nj() using the
microbenchmark package for robust timing:
library(microbenchmark)
# Create a 500-taxon distance matrix
set.seed(42)
n <- 500
d <- matrix(runif(n*n), n, n)
d <- (d + t(d)) / 2
diag(d) <- 0
rownames(d) <- colnames(d) <- paste0("t", 1:n)
# Run each method 10 times
mb <- microbenchmark(
`ape::nj()` = ape::nj(d),
`fast_nj(rapidnj)` = fast_nj(d, method = "rapidnj"),
`fast_nj(bionj)` = fast_nj(d, method = "bionj"),
times = 10
)
print(mb)
#> Unit: milliseconds
#> expr min lq mean median uq max neval
#> ape::nj() 74.27749 75.72017 77.10697 76.36930 78.03949 82.53536 10
#> fast_nj(rapidnj) 26.34244 26.81659 28.09161 28.19222 29.14561 30.58926 10
#> fast_nj(bionj) 19.24839 19.31572 19.99373 19.50370 20.84758 21.14330 10
# Calculate and display speedup
summary_mb <- summary(mb)
ape_median <- summary_mb[summary_mb$expr == "ape::nj()", "median"]
rapidnj_median <- summary_mb[summary_mb$expr == "fast_nj(rapidnj)", "median"]
cat(sprintf("\nSpeedup (median): %.1fx faster\n", ape_median / rapidnj_median))
#>
#> Speedup (median): 2.7x fasterScaling Behavior
The speedup increases dramatically with dataset size. Here are typical results:
| Taxa | ape::nj() | fast_nj() | Speedup |
|---|---|---|---|
| 100 | 1 ms | 1 ms | ~1x |
| 500 | 70 ms | 17 ms | ~4x |
| 1,000 | 550 ms | 70 ms | ~8x |
| 2,000 | 4.5 s | 250 ms | ~18x |
| 5,000 | 70 s | 1.5 s | ~47x |
For datasets with thousands of taxa, phylobuild makes neighbor-joining practical.
Working with Real Data
Integration with ape
phylobuild trees are fully compatible with the ape ecosystem:
data(primates)
tree <- fast_nj(primates)
# Standard ape operations work
Ntip(tree)
#> [1] 12
Nnode(tree)
#> [1] 10
is.rooted(tree)
#> [1] FALSE
# Tree manipulation
tree_rooted <- root(tree, outgroup = "Tarsier")
plot(tree_rooted)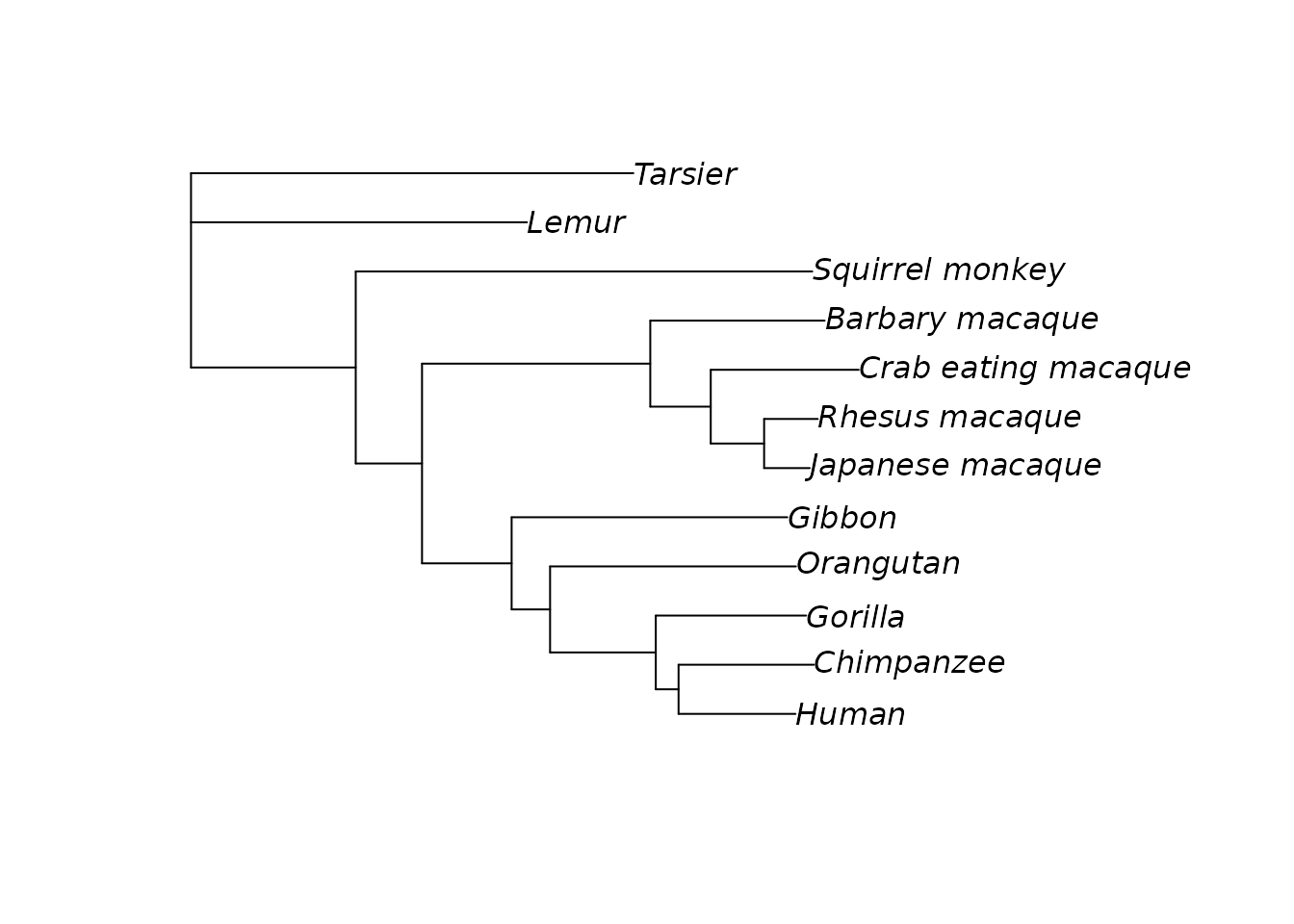
Tips for Large Datasets
- Use RapidNJ (the default) - it’s optimized for large matrices
- Monitor memory - distance matrices grow as O(n²)
- Consider subsampling - for very large datasets, subsample first
-
Save intermediate results - write trees to files
with
write.tree()
# For very large trees
tree <- fast_nj(huge_distance_matrix, method = "rapidnj")
write.tree(tree, "my_tree.nwk")References
- Simonsen M, Mailund T, Pedersen CNS (2011). “Inference of Large Phylogenies using Neighbour-Joining.” Communications in Computer and Information Science, 127, 334-344.
- Saitou N, Nei M (1987). “The neighbor-joining method: a new method for reconstructing phylogenetic trees.” Molecular Biology and Evolution, 4(4), 406-425.
- Gascuel O (1997). “BIONJ: An Improved Version of the NJ Algorithm Based on a Simple Model of Sequence Data.” Molecular Biology and Evolution.
Session Info
sessionInfo()
#> R version 4.5.2 (2025-10-31)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.3 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] microbenchmark_1.5.0 ape_5.8-1 phylobuild_0.1.0
#>
#> loaded via a namespace (and not attached):
#> [1] nlme_3.1-168 cli_3.6.5 knitr_1.51 rlang_1.1.7
#> [5] xfun_0.56 textshaping_1.0.4 jsonlite_2.0.0 htmltools_0.5.9
#> [9] ragg_1.5.0 sass_0.4.10 rmarkdown_2.30 grid_4.5.2
#> [13] evaluate_1.0.5 jquerylib_0.1.4 fastmap_1.2.0 yaml_2.3.12
#> [17] lifecycle_1.0.5 compiler_4.5.2 fs_1.6.6 Rcpp_1.1.1
#> [21] systemfonts_1.3.1 lattice_0.22-7 digest_0.6.39 R6_2.6.1
#> [25] parallel_4.5.2 bslib_0.10.0 tools_4.5.2 pkgdown_2.2.0
#> [29] cachem_1.1.0 desc_1.4.3